I. Fogalmak
In vitro diagnosztikai orvostechnikai eszköz: minden olyan orvostechnikai eszköz, amelyet – mint reagens, reagensszármazék, kalibráló és kontrollanyag, diagnosztikai kit, készülék, berendezés, gép, szoftver vagy rendszer – önmagában vagy más eszközzel együttesen alkalmazva a gyártó az emberi szervezetből származó minták – ideértve a vér- és a szövetadományozást is – kizárólag vagy elsősorban azzal a céllal történő in vitro vizsgálata céljából történő használatra szán, hogy információt szolgáltasson a következők bármelyikéről vagy a következő célok bármelyike érdekében:
- fiziológiai vagy patológiai folyamatról vagy állapotról,
- veleszületett testi vagy szellemi károsodásokról,
- adott kóros állapotra vagy betegségre való hajlamról,
- a potenciális recipiensek biztonságának és kompatibilitásának megítélése céljából,
- egy kezelésre adott válasznak vagy az általa kiváltott reakcióknak az előrejelzése céljából,
- terápiás intézkedések meghatározása vagy figyelemmel kísérése céljából.
A minták befogadására szolgáló tartályok in vitro diagnosztikai orvostechnikai eszköznek tekintendők.
Klinikai bizonyíték: valamely eszközhöz tartozó klinikai adatok és az eszköz teljesítőképesség-értékelésének eredményei, amelyek kellő mennyiségben és minőségben állnak rendelkezésre annak megalapozott eldöntéséhez, hogy a gyártó által előírt rendeltetés szerint használva az eszköz biztonságos-e és elérhető(k)-e vele a célzott klinikai előny(ök);
Klinikai előny: egy adott eszköznek a funkciójához (azaz például betegek szűréséhez, megfigyeléséhez, diagnosztizálásához vagy diagnosztizálásának elősegítéséhez) kapcsolódóan kifejtett kedvező hatása, illetve a betegellátásra vagy a közegészségre gyakorolt kedvező hatása;
Egy analit tudományos érvényessége: egy analit kapcsolata egy klinikai vagy fiziológiás állapottal;
Az eszköz teljesítőképessége: egy eszköz arra való alkalmassága, hogy teljesítse a gyártó állítása szerinti rendeltetését. A teljesítőképesség az e rendeltetést alátámasztó analitikai és adott esetben klinikai teljesítőképességből áll;
Analitikai teljesítőképesség: egy eszköz arra való alkalmassága, hogy egy bizonyos analitot helyesen felismerjen vagy mérjen;
Klinikai teljesítőképesség: egy eszköz arra való alkalmassága, hogy olyan eredményeket szolgáltasson, amelyek a célcsoportra és a célfelhasználókra nézve bizonyos klinikai állapottal vagy fiziológiás vagy patológiás folyamattal vagy állapottal korrelációt mutatnak;
Teljesítőképesség-vizsgálat: egy eszköz analitikai vagy klinikai teljesítőképességének megállapítása vagy bizonyítása érdekében végzett vizsgálat;
Teljesítőképesség-vizsgálati terv: olyan dokumentum, amely leírja egy adott teljesítőképesség-vizsgálat indokait, célkitűzéseit, elrendezését, módszertanát, nyomon követését, statisztikai megfontolásait, szervezését és lefolytatását;
Teljesítőképesség-értékelés: egy eszköz tudományos érvényességét, valamint analitikai, és adott esetben klinikai teljesítőképességét megállapító vagy bizonyító adatok értékelése és elemzése;
Teljesítőképesség-vizsgálatra szánt eszköz: olyan eszköz, amelyet a gyártó teljesítőképesség-vizsgálatban történő felhasználásra szán. Azok az eszközök, amelyeket kutatási célú felhasználásra szánnak, és nincs orvosi célú rendeltetésük, nem tekintendők teljesítőképesség-vizsgálatra szánt eszköznek;
Beavatkozással járó klinikai teljesítőképesség-vizsgálat: olyan klinikai teljesítőképesség-vizsgálat, amelynek esetében a vizsgálati eredmények hatással lehetnek a betegellátással kapcsolatos döntésekre és/vagy fel lehet azokat használni a kezelés irányának meghatározásához;
Vizsgálati alany: az az egyén, aki részt vesz egy teljesítőképesség-vizsgálatban oly módon, hogy a tőle származó mintán/mintákon teljesítőképesség-vizsgálatra szánt eszközzel és/vagy kontrolleszközzel in vitro vizsgálatot végeznek;
Vizsgáló: az az egyén, aki a teljesítőképesség-vizsgálatok helyszínén a teljesítőképesség-vizsgálat elvégzéséért felelős;
Megbízó: minden olyan személy, vállalkozás, intézmény vagy szervezet, aki vagy amely a teljesítőképességvizsgálat kezdeményezéséért, irányításáért és finanszírozásának meghatározásáért felelősséget vállal;
Tájékoztatáson alapuló beleegyező nyilatkozat: a teljesítőképesség-vizsgálat minden olyan szempontjáról történő értesülést követően, amely a vizsgálati alanynak a teljesítőképesség-vizsgálatban való részvételéről meghozandó döntése szempontjából meghatározó, a vizsgálati alany általi szabad és önkéntes kifejezése annak, hogy részt kíván venni az adott teljesítőképesség-vizsgálatban, illetve a kiskorúak és a korlátozottan cselekvőképes és cselekvőképtelen vizsgálati alanyok esetében a törvényes képviselőjük engedélye vagy beleegyezése a teljesítőképesség-vizsgálatba való bevonásukra vonatkozóan;
Etikai bizottság: valamely tagállamban a nemzeti joggal összhangban létrehozott független testület, amely felhatalmazással rendelkezik arra, hogy e rendelet alkalmazásában véleményt nyilvánítson, mégpedig laikus személyek, különösen betegek és betegszervezetek véleményének a figyelembevételével. Magyarországon ez az Egészségügyi Tudományos Tanács Tudományos és Kutatásetikai Bizottság (ETT TUKEB). Hozzájáruló véleményét a vizsgálati kérelemhez csatolni kell, ez feltétele a vizsgálati engedély kiadásának. Az etikai vélemény iránti kérelem, a vélemény kiállítására irányuló eljárás részleteire, illetve díjának mértékével kapcsolatban keresse fel az ETT TUKEB hivatalos honlapját: https://ett.aeek.hu/tukeb/
Nemkívánatos esemény: egy teljesítőképesség-vizsgálattal összefüggésben a vizsgálati alanyok, felhasználók vagy más egyének egészségi állapotában bekövetkező minden kedvezőtlen változás, betegellátással kapcsolatos helytelen döntés, előre nem látható megbetegedés vagy sérülés vagy kedvezőtlen klinikai tünet, ideértve a rendellenes laboratóriumi eredményeket is, függetlenül attól, hogy ezek a teljesítőképesség-vizsgálatra szánt eszközzel kapcsolatosak-e vagy sem;
Súlyos nemkívánatos esemény: minden olyan nemkívánatos esemény, amely a következők bármelyikéhez vezetett:
- olyan betegellátással kapcsolatos döntés, amelynek következtében a vizsgált egyén életét veszti vagy közvetlen életveszélybe kerül, vagy a vizsgált személy utódja életét veszti,
- halál,
- a vizsgált egyén vagy a vizsgált adományok vagy anyagok recipiense egészségi állapotának súlyos romlása, ami a következők bármelyikéhez vezetett:
- életet veszélyeztető megbetegedés vagy sérülés;
- az anatómiai struktúrák vagy élettani funkciók tartós károsodása;
- kórházi ápolás vagy a beteg kórházi ápolásának meghosszabbodása,
- életet veszélyeztető megbetegedés vagy sérülés megelőzése érdekében, vagy az anatómiai struktúrák vagy élettani funkciók tartós károsodásának megelőzése érdekében végrehajtott orvosi vagy sebészeti beavatkozás;
- krónikus betegség,
- magzati distressz, magzati halál, veleszületett testi vagy szellemi károsodás, vagy születési rendellenesség
Forgalomba hozatal utáni felügyelet: a gyártók által más gazdasági szereplőkkel együttműködésben végzett mindazon tevékenységek, amelyek célja az általuk forgalomba hozott, forgalmazott vagy használatba adott eszközeikkel kapcsolatban szerzett tapasztalatok proaktív módon történő összegzését és felülvizsgálatát lehetővé tevő szisztematikus eljárás kialakítása és napra készen tartása annak érdekében, hogy azonosítani lehessen minden olyan esetet, amikor haladéktalanul korrekciós vagy megelőző intézkedés alkalmazására van szükség;
Váratlan esemény: a forgalmazott eszköz tulajdonságaiban vagy teljesítőképességében bekövetkezett rendellenes működés vagy romlás – ideértve az ergonómiai jellemzőkből adódó felhasználási hibát is –, valamint a gyártó által szolgáltatott információk helytelensége és minden olyan ártalom, amely az eszköz által szolgáltatott információk vagy eredmény(ek) alapján meghozott orvosi döntés, elvégzett vagy el nem végzett beavatkozás következménye;
Súlyos váratlan esemény: minden olyan váratlan esemény, amely közvetetten vagy közvetlenül a következőkhöz vezetett, vezethetett volna vagy vezethet:
- a beteg, a felhasználó vagy más egyén halála,
- a beteg, a felhasználó vagy más egyén egészségi állapotának súlyos átmeneti vagy tartós romlása,
- súlyos közegészségügyi kockázat;
Súlyos közegészségügyi kockázat: minden olyan esemény, amely magában hordozhatja a halálnak, egy egyén egészségi állapota súlyos romlásának vagy súlyos betegség bekövetkezésének a közvetlen kockázatát, és amelynek az esetében gyors korrekciós intézkedések válhatnak szükségessé, valamint amely jelentős arányban okozhat emberi megbetegedést vagy elhalálozást, vagy amely szokatlan vagy váratlan az adott helyszínen és időpontban.
II. Vonatkozó jogszabályok
Jogszabályok, melyek az IVD eszközök teljesítőképesség-vizsgálatával összefüggenek:
- az egészségügyről szóló 1997. évi CLIV. törvény;
- az in vitro diagnosztikai orvostechnikai eszközökről szóló EU 2017/746 rendelet (IVDR);
- az in vitro diagnosztikai orvostechnikai eszközökről szóló 8/2003. (III. 13.) ESZCSM rendelet;
- az orvostechnikai eszközök klinikai vizsgálatáról szóló 33/2009. (X. 20.) EüM rendelet;
- az emberen végzett orvostudományi kutatások, az emberi felhasználásra kerülő vizsgálati készítmények klinikai vizsgálata, valamint az emberen történő alkalmazásra szolgáló, klinikai vizsgálatra szánt orvostechnikai eszközök klinikai vizsgálata engedélyezési eljárásának szabályairól szóló 235/2009. (X. 20.) Korm. rendelet;
- az emberen végzett orvostudományi kutatásokról szóló 23/2002. (V. 9.) EüM rendelet;
- a népjóléti ágazatba tartozó egyes államigazgatási eljárásokért és igazgatási jellegű szolgáltatásokért fizetendő díjakról szóló 50/1996. (XII. 27.) NM rendelet.
III. Általános tudnivalók
Az IVD eszközök teljesítőképesség-értékelését az (EU) 2017/746 rendelete (továbbiakban IVDR) 56. cikke írja elő a gyártók részére. A teljesítőképesség-értékelés célja, hogy kellő klinikai bizonyítékot szolgáltasson arról, hogy az adott IVD eszköz megfelel-e a biztonságosságra és a teljesítőképességre vonatkozó követelményeknek, és ezek alapján kell értékelni az interferenciákat, keresztreakciókat, valamint az eszköz használatával járó előny-kockázat arány elfogadhatóságát.
A teljestőképesség értékelés során az alábbiakat kell igazolni:
- tudományos érvényesség;
- analitikai teljesítőképesség;
- klinikai teljesítőképesség.
A három elem értékeléséből származó eredmények szolgáltatják a kellő klinikai bizonyítékot.
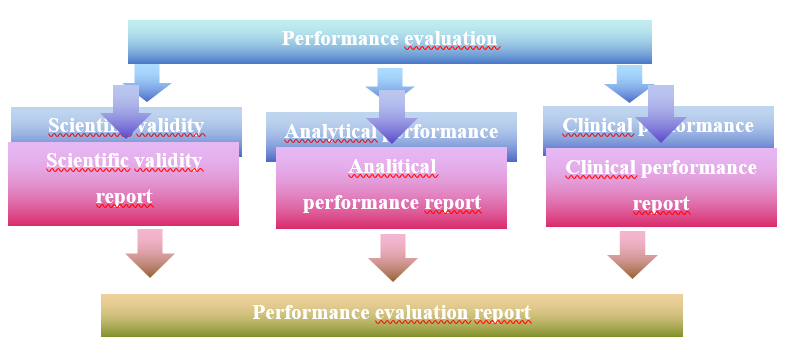
A teljesítőképesség-értékelés elvégzésének folyamata
A teljesítőképesség-értékelés egy dokumentált, teljesítőképesség-értékelési tervet követő folyamatos tevékenység. Megvalósításához a gyártónak el kell készítenie a teljesítőképesség-értékelési tervet az IVDR XIII. melléklet 1.1 pontja szerint. Ezt követően el kell végeznie a tudományos érvényesség igazolását (IVDR XIII. melléklet 1.2.1.), az analitikai teljesítőképesség igazolását (IVDR XIII. melléklet 1.2.2.) és a klinikai teljesítőképesség igazolását (IVDR XIII. melléklet 1.2.3.). Mindhárom lépést egy-egy jelentésben kell igazolni. A három jelentésből pedig el kell készíteni a teljesítőképesség-értékelési jelentést (PER) (IVDR XIII. melléklet A. rész 1.3.2. pont). A folyamatot az alábbi ábra szemlélteti:
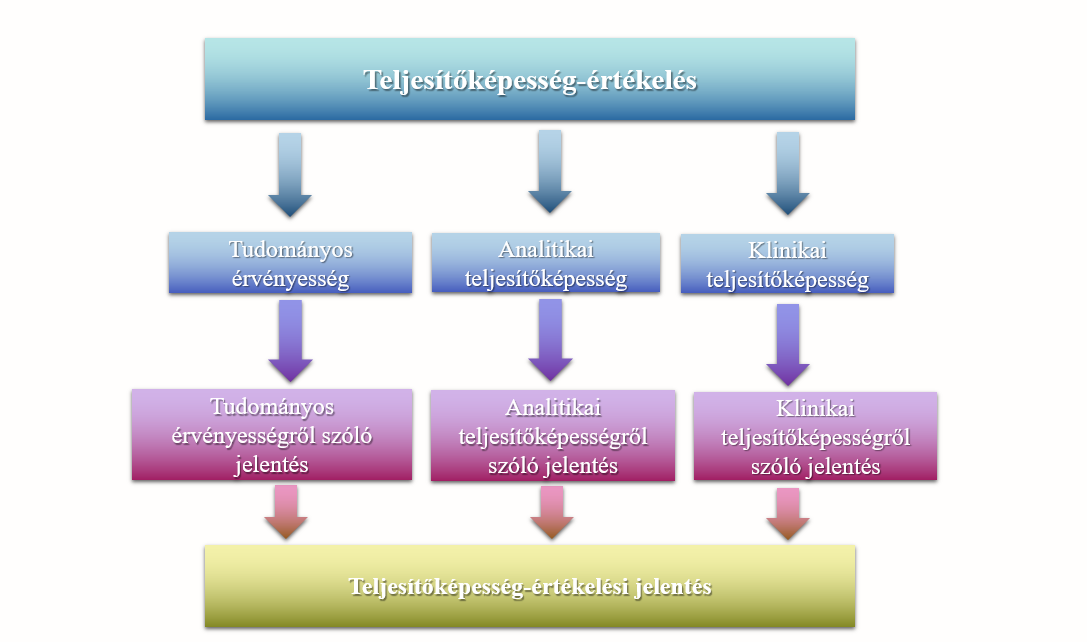
- ábra: A teljesítőképesség-értékelés folyamata
A teljesítőképesség-értékelés a műszaki dokumentáció része.
A teljesítőképesség-értékelést és annak dokumentációját az adott eszköz teljes életciklusa során aktualizálni kell, melynek alapja a forgalomba hozatal utáni felügyeleti rendszer és a hozzá tartozó terv, ami a végrehajtását szolgálja. (IVDR 78-79. cikk és a III. melléklet 1. pontja).
A forgalomba hozatal utáni felügyeleti rendszeren keresztül számos adat gyűjthető, melyet az IVDR 78. cikk (3) bekezdése sorol fel. Kiemelkedő jelentőségű lehet a teljesítőképesség forgalomba hozatal utáni nyomon követése (XIII. melléklet B. rész szerint) és az erre vonatkozó terv, melyben akár egy a teljesítőképesség forgalomba hozatal utáni nyomon követését szolgáló vizsgálat (PMPF study) is szerepelhet.
A C és D. osztályba sorolt eszközökre elkészített teljesítőképesség-értékelési jelentést (PER) szükség szerint, de legalább évente aktualizálni kell (időszakos eszközbiztonsági jelentés – IVDR 81. cikk). Ehhez hasonlóan az A és B osztályba sorolt eszközök esetében forgalomba hozatal utáni felügyeleti jelentést kell készíteni rendszeresen a gyártóknak (IVDR 80. cikk). Továbbá a C. és D. osztályba sorolt eszközökre az IVDR 29. cikke értelmében el kell készíteni a biztonságosságra és teljesítőképességre vonatkozó összefoglalót (SSCP), melyet úgy kell megszövegezni, hogy a célfelhasználók részére is egyértelmű legyen, valamint nyilvánosan hozzáférhetővé kell tenni az EUDAMED-en keresztül. Az EUDAMED működőképessé válásáig a gyártó a honlapján, vagy más, nyilvánosan elérhető, internetes felületen történő közzététellel tesz eleget.
IV. Teljesítőképeség-értékelés elvégzésének módjai
1. A teljesítőképesség-vizsgálatokra vonatkozó általános követelmények
- a teljesítőképesség-vizsgálatokat az eszköz rendes használati feltételeihez hasonló körülmények között kell végezni;
- a gyártó biztosítja, hogy a teljesítőképesség-vizsgálatra szánt eszköz megfelel az I. mellékletben említett, a biztonságosságra és a teljesítőképességre vonatkozó általános követelményeknek, kivéve a teljesítőképesség-vizsgálatban meghatározott szempontokat;
- a teljesítőképesség-vizsgálatokat úgy kell megtervezni és végrehajtani, hogy megvalósuljon és minden más érdekkel szemben elsőbbséget élvezzen az ilyen teljesítőképesség-vizsgálatokban részt vevő személyek jogainak, biztonságának, méltóságának és jóllétének a védelme, valamint, hogy a vizsgálatokból származó klinikai adatok tudományosan érvényesek, megbízhatók és megalapozottak legyenek.
- A teljesítőképesség-vizsgálatokat – a maradványmintákat használó teljesítőképesség-vizsgálatokat is beleértve – az adatvédelemre vonatkozó hatályos jognak megfelelően kell végezni.
- Az IVDR rendelet alkalmazásának időpontja 2022. május 26. Azon teljesítőképesség vizsgálatok esetében, melyeket ezen időpont előtt benyújtottak, a 98/79/EK irányelv (IVDD) és az ezt honosító, az in vitro diagnosztikai orvostechnikai eszközökről szóló 8/2003. (III.13.) ESzCsM rendelet előírásai szerint folytathatók.
- május 26-át követően már csak az IVDR-nek megfelelően lehet teljesítőképesség vizsgálatot engedélyeztetni, lefolytatni.
2. Teljesítőképesség-értékelés jól megalapozott (well-established) eszköz esetén
Amennyiben az eszköz régóta forgalomban van és a gyártónak minden szükséges információ rendelkezésre áll, akkor elkészíti az előbbiekben ismertetett teljesítőképesség-értékelési tervet, majd a tudományos érvényességről szóló jelentést, az analitikai teljesítőképességről szóló jelentést és a klinikai teljesítőképességről szóló jelentést, ezt követően pedig elkészíti – ezek összegzéseként – a teljesítőképesség-értékelési jelentést. A teljesítőképesség-értékelési dokumentációnak az adott eszköz műszaki dokumentációjának részét kell képeznie.
Az IVDR 56. cikkének (4) pontja értelmében a XIII. melléklet A. részének 2. pontja szerinti klinikai teljesítőképesség-vizsgálatok elvégzésétől csak abban az esetben lehet eltekinteni, ha a klinikai teljesítőképességre vonatkozó adatok egyéb forrásainak igénybevétele kellőképpen indokolt.
3. Hatósági bevonást igénylő teljesítőképesség vizsgálatok
3.1. Kérelem/bejelentés benyújtásának feltételei és előzményei
- Amennyiben az adott teljesítőképesség-vizsgálat megbízója nincs letelepedve az Európai Unióban, gondoskodnia kell arról, hogy egy unión belül letelepedett természetes vagy jogi személy lássa el az unión belül a jogi képviseletét (IVDR 58. cikk (4) bekezdés).
- A Magyarország területén székhellyel rendelkező, illetve a Magyarország területén székhellyel rendelkező meghatalmazott képviselővel rendelkező gyártónak, a Magyarország területén székhellyel rendelkező meghatalmazott képviselőnek az IVDR 28. cikk szerinti regisztrációnak eleget kell tennie, SRN-számmal rendelkeznie kell. A klinikai vizsgálatra szánt eszköz gyártójának minden esetben rendelkeznie kell SRN számmal https://ogyei.gov.hu/eudamed_actor_modul
- A teljesítőképesség-vizsgálat megbízójának kérelmet/bejelentést kell benyújtania a klinikai vizsgálat végzésének helye szerinti tagállam orvostechnikai eszköz hatóságához (NNGYK).
- A kérelmet/bejelentést az IVDR 69. cikkben említett elektronikus rendszeren keresztül (EUDAMED klinikai vizsgálat modul) kell benyújtani. Ameddig az EUDAMED „klinikai vizsgálatok modul”-ja nem üzemel, addig az NNGYK a kérelmeket e-ügyintézés* keretében fogadja.
- Az adott teljesítőképesség-vizsgálatra/IVD eszközös vizsgálatra vonatkozóan uniószerte egységes, egyedi azonosító számot kell generálnia az EUDAMED rendszernek (CIV-ID), amelyet minden esetben meg kell adni az adott klinikai vizsgálattal kapcsolatos kommunikáció során. Ameddig az EUDAMED „klinikai vizsgálatok modul”-ja nem üzemel, addig az NNGYK az EUDAMED2 rendszerben nyilvántartásba veszi a klinikai vizsgálatot és az ott kapott azonosítót megküldi a megbízónak.
*Az elektronikus ügyintézés és a bizalmi szolgáltatások általános szabályairól szóló 2015. évi CCXXII. törvény (e-ügyintézési tv.) értelmében 2018. január 1-től kötelező a gazdálkodó szervezetek számára az egyes állami szervekkel való elektronikus kapcsolattartás, valamint az ehhez szükséges hivatalos elérhetőségen történő dokumentumküldés/fogadás. Hivatalos ügy indításához segédlet az alábbi linken található: https://ogyei.gov.hu/eugyintezes
3.1.1. Pozitív etikai vélemény szerzése
A teljesítőképesség-értékelési vizsgálat az Egészségügyi Tudományos Tanács Tudományos és Kutatásetikai Bizottság (ETT TUKEB) által – az (EU) 2017/746 rendelet 58. cikk (3) alapján - kiadott támogató etikai véleményének birtokában kezdeményezhető az NNGYK-nál.
Az etikai véleménnyel kapcsolatosan kérjük, keresse az Egészségügyi Tudományos Tanács Tudományos és Kutatásetikai Bizottságot (ETT TUKEB) az alábbi elérhetőségeken:
Levelezési cím: Belügyminisztérium, 1903 Budapest, Pf.: 314.
Telefon: (+36 1) 795-1197 (+36 1) 795-1198
E-mail:
Honlap: https://ett.okfo.gov.hu/tukeb/
3.2. Bejelentésköteles teljesítőképesség vizsgálatok
3.2.1. Teljesítőképesség-vizsgálatok maradványminták felhasználásával
Az egészségügyről szóló 1997. évi CLIV. törvény 164/A. §. (10) pontja értelmében a bejelentésköteles teljesítőképesség-értékelési vizsgálatok a következők:
- az (EU) 2017/746 rendelet 58. cikk (1) bekezdése alá nem tartozó, in vitro diagnosztikai orvostechnikai eszközökkel végzett teljesítőképesség-vizsgálatok, (nem történik a teljesítőképesség-vizsgálat céljából célzott mintavétel, vagyis a teljesítőképesség-vizsgálat maradékminták felhasználásával történik);
- az (EU) 2017/746 rendelet 58. cikk (2) bekezdése szerinti, a kapcsolt diagnosztikumoknak a kizárólag maradványminták felhasználásával végzett teljesítőképesség-vizsgálata.
Az NNGYK a bejelentés megtételét követő 60 napon belül hatósági bizonyítványt állít ki.
3.2.2. Teljesítőképesség-vizsgálatok CE-jelöléssel ellátott eszközök esetében
Az IVDR 70. cikk (1) bekezdése szerinti vizsgálat esetén a megbízónak legkésőbb 30 nappal a vizsgálat megkezdését megelőzően be kell jelentenie az NNGYK részére a teljesítőképesség-vizsgálatot az EUDAMED-en keresztül.
A vizsgálat a 1997. évi CLIV. törvény 164/A. §. (10) pontja értelmében a bejelentésköteles, azonban a kérelem benyújtása során a 2.6. pontban ismertetett eljárásrend követendő. Amennyiben a benyújtott bejelentés hiányos, csak a teljes kiegészítéstől számított 30. napon kezdhető meg a vizsgálat a nyilvántartásba vételi igazolás birtokában. Felhívjuk figyelmét, hogy az Eütv. lehetőséget ad az NNGYK-nak, hogy az igazolást a hiánytalan bejelentés időpontjától számított 30 naptári napon belül állítsa ki.
IVDR 70. cikk (2) bekezdése szerinti vizsgálat esetén a kérelem benyújtásának módja a mintavétel módjától függ.
3.3. Engedélyköteles teljesítőképesség-értékelési vizsgálatok
Az engedélyköteles vizsgálatok körét az emberen végzett orvostudományi kutatások, az emberi felhasználásra kerülő vizsgálati készítmények klinikai vizsgálata, valamint az emberen történő alkalmazásra szolgáló, klinikai vizsgálatra szánt orvostechnikai eszközök klinikai vizsgálata engedélyezési eljárásának szabályairól szóló 235/2009. (X.20.) Korm. rendelet határozza meg. A fenti rendelet 30.§ (1) pontja értelmében engedélyköteles vizsgálatok a következők:
- az IVDR 58. cikk (1) bekezdése szerinti olyan teljesítőképesség-vizsgálatok,
- amelyek során kizárólag a teljesítőképesség-vizsgálat céljából kerül sor sebészeti invazív mintavételre;
- amelyek az IVDR 2. cikk 46. pontjában meghatározott, beavatkozással járó klinikai teljesítőképesség-vizsgálatok; vagy
- amelyek során a vizsgálat további invazív eljárásokkal vagy a vizsgálati alanyokra nézve egyéb kockázatokkal jár,
- az IVDR 58. cikk (2) bekezdése szerinti kapcsolt diagnosztikum nem maradványminták felhasználásával végzett vizsgálata,
- IVDR 70. cikk (2) bekezdése szerinti teljesítőképesség-vizsgálatok (amennyiben célzott mintavétel történik).
Az IVDR 58. cikk (1) bekezdése szerinti teljesítőképesség-értékelési vizsgálat az ETT TUKEB bizottságának az (EU) 2017/746 rendelet 58. cikk (5) bekezdés b) pontja szerint kiadott támogató etikai véleményének birtokában kérelmezhető.
A vizsgálatot az NNGYK engedélyezi és nyilvántartásba veszi a 235/2009. (X.20.) Korm. rendelet 32. § -nak megfelelően.
A teljesítőképesség-vizsgálatot csak akkor lehet lefolytatni, ha az IVDR 58. cikk (5) pontjában felsorolt feltételek mindegyike teljesül.
3.4. A hatósági eljárás folyamata
Felhívjuk figyelmüket, hogy a kérelem befogadására és elbírálására az IVDR 66. cikkének rendelkezései az irányadók.
- A teljesítőképesség-vizsgálat iránti kérelem beérkezését követően az NNGYK 10 napon belül értesíti a megbízót (ezt a határidőt az NNGYK 5 nappal meghosszabbíthatja), hogy a teljesítőképesség-vizsgálat az IVDR hatálya alá tartozik-e és a beérkezett kérelem hiánytalan-e.
- Ha az NNGYK megállapítja, hogy a teljesítőképesség-vizsgálat nem tartozik az IVDR hatálya alá, vagy a kérelem hiányos, tájékoztatja a megbízót és 10 napos határidőt állapít meg (ezt adott esetben az NNGYK kérelemre 20 nappal meghosszabbíthatja), hogy a megbízó a kérelemhez észrevételt tegyen, vagy azt kiegészítse. Az észrevételt és kiegészítést az EUDAMED-en/e-ügyintézésen keresztül kell megtenni.
- Amennyiben a megbízó a fenti határidőn belül nem tett észrevételt és/vagy a kérelmet nem egésztette ki, a kérelmet megszűntnek kell tekinteni. (eljárást megszüntető végzés kerül kiküldésre)
- Ha a megbízó úgy ítéli meg, hogy a kérelem az IVDR hatálya alá tartozik és/vagy az szerinte hiánytalan, de az NNGYK ezzel nem ért egyet a kérelmet elutasítottnak kell tekinteni (eljárást megszüntető végzés kerül kiküldésre). Az NNGYK jogorvoslati eljárást biztosít az eljárás megszüntetése esetén.
- Amennyiben a megbízó a fenti határidőn belül észrevételt tett, ill. kiegészítést tett, az NNGYK annak kézhezvételétől számított 5 napon belül (ezt a határidőt az NNGYK 5 nappal meghosszabbíthatja) értesíti a megbízót, hogy a teljesítőképesség-vizsgálat az IVDR hatálya alá tartozik-e és a beérkezett kérelem hiánytalan-e. (Ez a kérelem validálásának közlése.)
- A határidők minden esetben naptári napokban számolandók!
3.4.1. A kérelem validálása
- Az a dátum, amikor a megbízót az NNGYK értesíti, hogy a kérelem az IVDR hatálya alá tartozik és a kérelem hiánytalan.
- A kérelem validálása után kezdi meg az NNGYK az IVDR 67. cikkében meghatározott értékelést, mely alatt a hatóság felszólíthatja a megbízót kiegészítő információk benyújtására. Az NNGYK a validálás dátumától számított 45 napon belül – az értékelési eljárás végén – értesíti a megbízót az engedélyről. A tagállam szakértői konzultációk céljából további 20 nappal meghosszabbíthatja az értékelésre adott időtartamot.
A megbízó az alábbi körülmények között kezdheti el a teljesítőképesség vizsgálatot:
- Az IVDR 58. cikk (1) bekezdésének a) pontja szerint végzett olyan teljesítőképesség-vizsgálat esetén, amely során a mintagyűjtés nem jár jelentős klinikai kockázattal a vizsgálati alanyokra nézve a vizsgálat a kérelem validálását követően kezdhető meg. (lsd. a példákat a 2. sz. mellékletben)
- Az alábbi teljesítőképesség-vizsgálatokat csak az NNGYK által kiadott engedély véglegessé válását követően lehet megkezdeni:
- az IVDR 58. cikk (1) bekezdése szerinti olyan teljesítőképesség-vizsgálatok,
- amelyek során kizárólag a teljesítőképesség-vizsgálat céljából kerül sor sebészeti invazív mintavételre és ez jelentős kockázattal jár a vizsgálati alanyokra nézve; lsd. 2. sz. melléklet.
- amelyek az IVDR 2. cikk 46. pontjában meghatározott, beavatkozással járó klinikai teljesítőképesség-vizsgálatok; vagy
- amelyek során a vizsgálat további invazív eljárásokkal vagy a vizsgálati alanyokra nézve egyéb kockázatokkal jár (lsd. a példákat a 2. sz. mellékletben),
- az IVDR 58. cikk (2) bekezdése szerinti kapcsolt diagnosztikum nem maradványminták felhasználásával végzett vizsgálata.
- az IVDR 58. cikk (1) bekezdése szerinti olyan teljesítőképesség-vizsgálatok,
V. A benyújtandó dokumentumok listája
A dokumentumokat egy mappába, összezippelve a Mellékletek/Annexes dokumentumnak megfelelően sorszámozva kérjük benyújtani!
|
Dokumentum |
Engedélyköteles IVDR 58. cikk, 70. cikk (2) |
Bejelentésköteles IVDR 70.cikk (1) CE-jelöléssel ellátott eszköz esetén |
Bejelentésköteles 1997. évi CLIV tv.164/A. §.(10). |
Megjegyzés |
|
Mellékletek/Annexes dokumentum |
+ |
+ |
+ |
Keressék honlapunkon! |
|
Kísérőlevél |
+ |
+ |
+ |
szabadszavas levél az NNGYK Orvostechnikai Főosztálynak címezve, amiben kérik a benyújtott dokumentumok elbírálását |
|
Az igazgatási-szolgáltatási díj megfizetését igazoló bizonylat |
+ |
+ |
+ |
|
|
A teljesítőképesség-értékelésben részt vevő eszközök teljes listája Excelben |
+ |
+ |
+ |
|
|
Az EU-s jogi képviselő meghatalmazása, ha a szponzor EU-n kívüli |
+ |
+ |
+ |
akkor, ha a szponzor székhelye nem az EU-ban van |
|
Teljesítőképesség-értékelés bejelentés/kérelem formanyomtatvány |
+ |
+ |
+ |
Keressék honlapunkon!; MDCG 2022-19 https://ogyei.gov.hu/dynamic/performance_study_application_form_hu.pdf |
|
Teljesítőképesség-értékelési terv (Performance Evaluation Plan-PEP) |
+ |
+ |
+ |
IVDR XIII. melléklet A. rész 1.1 szerint (tudományos érvényességre, analitikai teljesítőképességre, klinikai teljesítőképességre vonatkozó terv); MDCG 2022-2 |
|
Klinikai teljesítőképesség-vizsgálati terv (Clinical performance study plan-CPSP) |
+ |
+ |
+ |
IVDR XIII. melléklet A. rész 2.3.2. szerint |
|
A vizsgáló részére összeállított ismertető (Investigator's Brochure-IB). Tartalmi elemei: |
+ |
+ |
- |
A vizsgálói kézikönyv tartalmára vonatkozó |
|
• az eszköz azonosítása és leírása, ideértve a rendeltetéssel, a kockázati osztályba sorolással és a VIII. melléklet szerinti alkalmazandó osztályozási szabályokkal, valamint az eszkök kialakításával és gyártásával kapcsolatos információkat is, továbbá hivatkozás az eszköz korábbi és hasonló generációira |
||||
|
• a gyártó útmutatói az üzembe helyezéshez, a karbantartáshoz, a higiéniai előírások betartásához és a használathoz, ideértve a tárolási és a kezelési követelményeket is, továbbá – amennyiben az információ rendelkezésre áll – a címkén feltüntetendő információk és a forgalomba hozatalkor az eszközhöz mellékelendő használati útmutató. Az ismertetőnek ezenkívül a szükséges releváns képzéssel kapcsolatos információkat is tartalmaznia kell; |
||||
|
• analitikai teljesítőképesség; |
||||
|
• meglévő klinikai adatok |
||||
|
• előny-kockázat elemzés |
||||
|
• az emberi, állati vagy mikrobiális eredetű szöveteket, sejteket és anyagokat tartalmazó eszközök esetében részletes információk a szövetekről, sejtekről és anyagokról, valamint a biztonságosságra és a teljesítőképességre vonatkozó releváns általános követelményeknek való megfelelőségről, továbbá a szövetekkel, sejtekkel és anyagokkal kapcsolatos egyedi kockázatkezelés |
||||
|
• a biztonságosságra és teljesítőképességre vonatkozó általános követelmények teljesítését részletező lista, beleértve a részben vagy egészben alkalmazott szabványokat és egységes előírásokat, valamint a biztonságosságra és teljesítőképességre vonatkozó releváns általános követelmények teljesítésére szolgáló megoldások leírását, amennyiben ezeket a szabványokat és egységes előírásokat nem, vagy csak részben teljesítették, illetve azok hiányoznak |
||||
|
• a teljesítőképesség-vizsgálat folyamán alkalmazott klinikai eljárások és diagnosztikai tesztek részletes leírása, és különösen a standard klinikai gyakorlattól való eltérésre vonatkozó információk. |
||||
|
Az eszköz biztonságosságára és teljesítőképességére vonatkozó lista (GSPR list) |
+ |
+ |
+ |
lásd IVDR XIV. melléklet 4.1 |
|
Pozitív etikai szakvélemény |
+ |
+ |
+ |
|
|
A vizsgálati alanyok sérülés esetére szóló biztosításának vagy kártérítésre való jogosultságának az igazolása |
+ |
+ |
- |
lásd IVDR XIV. melléklet 4.3 |
|
A beleegyező nyilatkozat beszerzéséhez használandó dokumentumok, beleértve a betegtájékoztatót és a beleegyező nyilatkozatot tartalmazó dokumentumot |
+ |
+ |
- |
ICF (lásd IVDR XIV. melléklet 4.4) |
|
A személyes adatok védelmére és bizalmas kezelésére alkalmazandó szabályoknak való megfelelést célzó intézkedések leírása |
+ |
+ |
+ |
lásd IVDR XIV. melléklet 4.5 |
|
A rendelkezésre álló műszaki dokumentáció listája és nyilatkozat a hatóság felé, hogy kérésre benyújtja a dokumentációt |
+ |
- |
- |
lásd IVDR XIV. melléklet 4.6 |
|
Használati utasítás/ gépkönyv angol és magyar nyelven |
+ |
+ |
+ |
|
|
Gyártói megfelelőségi nyilatkozat (DoC) |
- |
+ |
- |
|
|
Címkeminta |
+ |
+ |
+ |
|
|
Az eszköz osztályba sorolásától függően az érvényes CE-tanúsítvány vagy a regisztrációt igazoló dokumentum |
- |
+ |
- |
|
|
A komparátor eszköz dokumentációja (DoC, CE-tanúsítvány, használati utasítás) |
+ |
+ |
+ |
referenciaeszköz dokumentációja |
|
Teljesítőképesség forgalomba hozatal utáni nyomonkövetési terve |
- |
+ |
+ (CE jelölt eszköz vizsgálata esetén) |
IVDR XIII. melléklet B. rész |
|
Egyéb tagállamok döntései |
+ |
+ |
+ |
|
|
Vizsgálók önéletrajza |
+ |
+ |
- |
|
|
Helsinki nyilatkozat |
+ |
+ |
- |
|
|
Vizsgálóhely befogadó nyilatkozata |
+ |
+ |
- |
|
|
Vizsgálóhelyek alkalmasságát igazoló nyilatkozat |
+ |
+ |
- |
|
|
Pénzügyi és egyéb rendelkezések |
+ |
+ |
+ |
VI. A teljesítőképesség-vizsgálat módosítása
A teljesítőképesség-vizsgálat jelentős módosításnak minősül, ha a vizsgálat módosítása valószínűleg jelentős hatást gyakorol a vizsgálati alanyok biztonságára, egészségére vagy jogaira, illetve a vizsgálat révén nyert adatok megbízhatóságára vagy megalapozottságára.
A vizsgálat jelentős vagy lényeges módosításnak minősül az IVDR 58. cikk (1) bekezdés szerinti teljesítőképesség-vizsgálatok esetében (235/2009. (X.20.) Korm. rendelet 35. §), ha:
- a módosítás az eszköz alkalmazási céljára, körülményeire vagy hatásmechanizmusára vonatkozik,
- a módosítás megváltoztathatja a vizsgálat elvégzését alátámasztó tudományos dokumentumok értelmezését,
- a módosítás a vizsgálók részére készített ismertetőt érinti,
- az addigi vizsgálati eredmények az írásos tájékoztató módosítását teszik szükségessé, vagy
- a módosítás hatással lehet a vizsgálati alanyok biztonságára, egészségére vagy jogaira.
A bejelentésköteles IVD-eszközökkel végzett vizsgálatok esetében a vizsgálati terv lényeges módosításának minősül (235/2009. (X.20.) Korm. rendelet 19. § (2) bekezdés) különösen, ha:
- a módosítás hatással lehet a vizsgálati alanyok biztonságára,
- a módosítás megváltoztathatja a beavatkozással nem járó vizsgálat elvégzését alátámasztó tudományos dokumentumok értelmezését,
- a módosítás a vizsgálók részére készített ismertetőt érinti,
- az addigi vizsgálati eredmények az írásos tájékoztató módosítását teszik szükségessé.
Felhívjuk figyelmüket, hogy a klinikai vizsgálatot engedélyező határozatban vagy hatósági bizonyítványban szereplő adatokban (pl. vizsgálati helyszín, vizsgálat vezető, megbízó adatai, bevont alanyok száma, vizsgálati idő stb.) történő változást és/vagy módosítást is be kell jelenteni az NNGYK felé.
1. Módosítás benyújtásának módja és ideje
Amennyiben a megbízó az IVD eszközzel végzett vizsgálatot oly módon szándékozik módosítani, hogy az jelentős módosításnak tekinthető (IVDR 71. cikk), illetve, ha a XIV. mellékletben említett dokumentáció bármely változásától számított egy héten belül naprakésszé kell tennie a vonatkozó adatokat, egyértelműen jelölnie kell a dokumentációra vonatkozó változást. Továbbá az EUDAMED „klinikai vizsgálatok modul”-ján keresztül ezen egy héten belül értesítenie kell a módosítás okairól és jellegéről az(oka)t a tagállamo(ka)t, ahol a klinikai vizsgálatot végzik vagy végezni fogják. Ameddig az EUDAMED „klinikai vizsgálatok modul”-ja nem üzemel, addig az NNGYK e-ügyintézés keretében fogadja a módosítási kérelmeket vagy bejelentéseket.
2. Kérelem és azzal benyújtandó dokumentumok
Az értesítés részeként a megbízónak csatolnia kell az IVDR XIV. mellékletében említett vonatkozó dokumentáció naprakész változatát az NNGYK honlapján megtalálható kérelem/bejelentés formanyomtatvány mellett. A vonatkozó dokumentációban egyértelműen meg kell jelölni a módosításokat. Az igazgatási szolgáltatási díj befizetéséről szóló bizonylat mellett az ETT TUKEB etikai véleménye a módosítási kérelem/bejelentés melléklete. Az etikai bizottság negatív, nem támogató vélemény esetén vizsgálat, annak engedélye/nyilvántartása nem módosítható.
3. Módosítást jóváhagyó hatósági eljárás
Az NNGYK az IVDR 67. cikkben megállapított eljárásnak megfelelően megvizsgálja minden esetben a klinikai vizsgálat jelentős módosítását.
4. Mikor kezdhető meg a módosítások szerinti klinikai vizsgálat?
A megbízó a klinikai vizsgálatot a módosításokkal legkorábban 38 nappal az EUDAMED-ben (jelenleg e-ügyintézés keretében) megküldött hiánytalan értesítést követően hajthatja végre, kivéve, ha:
- az a tagállam, ahol a teljesítőképesség-vizsgálatot végzik, vagy végezni kívánják, arról értesítette a megbízót, hogy az IVDR 67. cikk (4) bekezdése vagy a közegészséggel, a vizsgálati alanyok és a felhasználók biztonságával vagy egészségével, illetve a közrenddel kapcsolatos megfontolások alapján elutasította a módosítást,
- vagy az érintett tagállam etikai bizottsága olyan kedvezőtlen véleményt adott ki a teljesítőképesség-vizsgálat jelentős módosításával kapcsolatban, amely az adott tagállam jogának megfelelően a tagállam területének egészére érvényes.
A módosítási kérelemben érintett tagállam(ok) szakértői konzultációk céljából további hét (7) nappal meghosszabbíthatja/meghosszabbíthatják az IVDR 71. cikk (3) bekezdésben említett 38 napos időtartamot. Felhívjuk a figyelmet, hogy a nemzeti jogszabályok alapján a módosítások átvezetésére irányuló eljárás a hatóság részéről 60 napig eltarthat.
VII. Hatósági eljárások díja
Az egészségügyről szóló 1997. évi CLIV. törvény (Eütv.) 164/B. §-a szerint in vitro diagnosztikai orvostechnikai eszközökkel végzett teljesítőképesség-vizsgálat engedélyezési, illetve bejelentési eljárásáért igazgatási szolgáltatási díjat kell fizetni. Az in vitro diagnosztikai eszközzel végzett teljesítőképesség-értékelési vizsgálatok minden típusa esetén az igazgatási szolgáltatási díj megfizetését igazolni kell a kérelem/bejelentés benyújtásakor, mely a hatósági eljárás elindításának feltétele; díjfizetés elmulasztása esetén a kérelem/bejelentés visszautasításra kerül.
|
In vitro diagnosztikai orvostechnikai eszköz teljesítőképesség-vizsgálatának – ide nem értve az egészségügyről szóló 1997. évi CLIV. törvény 160/A. § (10) bekezdése szerinti teljesítőképesség-vizsgálatokat – engedélyezése, a vizsgálati terv jelentős módosításának engedélyezése |
243 000 Ft/ kérelem |
|
In vitro diagnosztikai orvostechnikai eszközzel végzett teljesítőképesség-vizsgálat bejelentése és a vizsgálati terv jelentős vagy lényeges módosításának bejelentése esetében |
99 000 Ft/ kérelem |
Pénzügyi adatok utaláshoz:
Név: Nemzeti Népegészségügyi és Gyógyszerészeti Központ
Adószám: 15598787-2-43
Bank: Magyar Államkincstár
Bank székhely: 1139 Budapest, Hungary, Váci út 71.
Bankszámlaszám: 10032000-00290438-00000000
IBAN: HU551003-2000-0029-0438-0000-0000
Kérjük az utalás során a közlemény rovatban feltüntetni az „orvostechnika” szó mellett a vizsgálat azonosítóját, vagy a vizsgálat címét megadni. Felhívjuk figyelmüket továbbá, hogy az NNGYK csak az igazgatási szolgáltatási díj befizetőjének tud számlát kiállítani, mellyel kapcsolatban az utalást indító adataira és email címére is szüksége van az NNGYK-nak.
VIII. Teljesítőképesség-vizsgálat megszakítása és befejezése
- Amennyiben a megbízó átmenetileg megszakít vagy idő előtt véglegesen leállít egy teljesítőképesség-vizsgálatot, 15 napon belül – elektronikus rendszeren keresztül – értesítenie kell az átmeneti megszakításról vagy idő előtti végleges leállításról azokat a tagállamokat, ahol sor került a klinikai vizsgálat átmeneti megszakítására vagy idő előtti leállítására.
- Amennyiben a megbízó biztonsági okokból szakította meg átmenetileg vagy állította le idő előtt véglegesen a teljesítőképesség-vizsgálatot, arról 24 órán belül értesítenie kell valamennyi olyan tagállamot, ahol az adott teljesítőképesség-vizsgálatot végzik.
- A teljesítőképesség-vizsgálat befejezésének az utolsó alany utolsó vizsgálatát kell tekinteni, kivéve, ha a teljesítőképesség-vizsgálati tervben egy másik időpontot határoztak meg a klinikai vizsgálat befejezéseként.
- A megbízónak minden olyan tagállamot, amelyben a teljesítőképesség-vizsgálatot végezték, értesítenie kell a teljesítőképesség-vizsgálatnak az adott tagállamban való befejezéséről is. Az értesítést a teljesítőképesség-vizsgálatnak az érintett tagállam vonatkozásában történő befejezését követő 15 napon belül kell megküldeni.
- Az egynél több tagállamban végzett vizsgálatok esetében a megbízónak értesítenie kell az összes tagállamot, amelyben a teljesítőképesség-vizsgálatot végezték a teljesítőképesség-vizsgálatnak az összes tagállamban való befejezéséről. Az értesítést a teljesítőképesség-vizsgálat befejezését követő 15 napon belül meg kell tenni.
- A megbízónak a teljesítőképesség-vizsgálat befejezését követő egy éven, vagy az idő előtti végleges leállítást, vagy átmeneti megszakítást követő három hónapon belül – a teljesítőképesség-vizsgálat eredményétől függetlenül – be kell nyújtania az IVDR XIII. melléklet A. részének 2.3.3. pontjában említett teljesítőképesség-vizsgálati jelentést azon tagállamok számára, amelyekben a teljesítőképesség-vizsgálatot végezték.
- A teljesítőképesség-vizsgálati jelentést a célfelhasználók számára is könnyen érthető módon megfogalmazott összefoglalónak kell kísérnie. A megbízónak mind a jelentést, mind pedig az összefoglalót az EUDAMED elektronikus rendszeren keresztül kell benyújtania.
- Amennyiben tudományos okok miatt nem lehetséges a teljesítőképesség-vizsgálati jelentésnek a vizsgálat befejezésétől számított egy éven belüli benyújtása, azt rendelkezésre állásakor a lehető leghamarabb be kell nyújtani. Ebben az esetben a XIII. melléklet A. részének 2.3.2. pontjában említett klinikai teljesítőképesség-vizsgálati tervben kell meghatározni, hogy mikor válnak elérhetővé a teljesítőképesség-vizsgálat eredményei, és ehhez indokolást kell fűzni.
- Ha az IVD eszköz IVDR 26. cikknek megfelelő regisztrációjára nem kerül sor az összefoglalónak és a teljesítőképesség-vizsgálati jelentésnek az elektronikus rendszerben történő rögzítésétől számított egy éven belül, akkor az összefoglalót és a jelentést az említett egy év leteltével nyilvánosan hozzáférhetővé kell tenni.
- Ameddig az EUDAMED „klinikai vizsgálatok modul”-ja nem üzemel, addig az NNGYK ezen dokumentumokat e-ügyintézés keretében fogadja. Kérjük a CIV-ID és az NNGYK irat iktatószámát feltüntetni a kísérő levélben és a dokumentumokon.
- Az NNGYK az IVD eszközökkel végzett vizsgálatokkal kapcsolatban hozzá benyújtott záró vizsgálati jelentését megküldi az ETT TUKEB részére.
- Kérjük a Teljesítőképesség-értékelési riportot (PER) e-ügyintézés keretében megküldeni szíveskedjenek az NNGYK-nak az IVDR 73. cikk szerinti tartalommal és időben. A tárgyban kérjük, tüntesse fel a következőket: „A teljesítőképesség-vizsgálat befejezéséről szóló értesítés, NNGYK által kiadott ügyiratszám” (használja a jóváhagyó levélben megadott ügyiratszámot).
IX. A teljesítőképesség-vizsgálat során előforduló nemkívánatos események rögzítése és bejelentése
A megbízónak az IVDR 76. cikk (2) bekezdése szerinti a teljesítőképesség-vizsgálatokkal kapcsolatban haladéktalanul be kell jelentenie az alábbi vigilancia-eseményeket azon tagállamoknak, ahol a klinikai vizsgálatot végzik:
- bármely súlyos nemkívánatos esemény, amelyek ok-okozati összefüggésben áll az eszközzel, a referenciaeszközzel vagy a vizsgálati eljárással, vagy amely esetében ilyen ok-okozati összefüggés észszerűen feltételezhető;
- bármely olyan eszközhiba, amely megfelelő intézkedés vagy beavatkozás hiányában, illetve kedvezőtlenebb körülmények között súlyos nemkívánatos eseményhez vezethetett volna;
- bármely, az előző két pontban említett eseményekkel kapcsolatos új megállapítás.
Ezen jelentéseket a megbízó az MDR 69. cikkben említett, az EUDAMED „klinikai vizsgálat modul” elektronikus rendszeren keresztül teheti meg. Magyarország esetében ezen jelentéseket az NNGYK az
A megbízónak be kell jelentenie a klinikai vizsgálatok során előforduló nemkívánatos eseményeket azon érintett tagállamoknak is, ahol a vizsgálatot végzik, valamint minden, olyan harmadik országokban (EU tagállamain kívül) előforduló eseményt, ahol az IVDR hatálya alá tartozó vizsgálatokra vonatkozó vizsgálati tervvel azonos vizsgálati terv alapján vizsgálatot végeznek.
FIGYELEM! Az IVDR 70. cikk (1) bekezdésében említett, azaz a teljesítőképesség forgalomba hozatal utáni nyomon követését szolgáló vizsgálat esetén a váratlan esemény jelentés az IVDR 82-85. cikkben és 86. cikkben foglaltak szerint történik, és az MDR 87. cikkében említett, az EUDAMED „vigilancia modul” elektronikus rendszeren keresztül kell megküldeni a hatóságnak. Az NNGYK az
Azonban ha a teljesítőképesség forgalomba hozatal utáni nyomon követését szolgáló vizsgálat (PMCF / és beavatkozással nem járó) vizsgálat során ok–okozati összefüggést állapítanak meg a súlyos nemkívánatos esemény és az azt megelőző PMCF-vizsgálati eljárás között, akkor az IVDR 76. cikket kell alkalmazni a vigilancia jelentésre és az EUDAMED „klinikai vizsgálat modul”-jába kell a jelentést megtenni, illetve az NNGYK az
A CE-jellel ellátott orvostechnikai eszközökkel (beleértve az IVD-eszközöket is) kapcsolatos váratlan esemény jelentésről tájékoztató és formanyomtatványok az alábbi linkeken érhetők el az NNGYK honlapján: