1. A teljesítőképesség-vizsgálatokra vonatkozó általános követelmények
- a teljesítőképesség-vizsgálatokat az eszköz rendes használati feltételeihez hasonló körülmények között kell végezni;
- a gyártó biztosítja, hogy a teljesítőképesség-vizsgálatra szánt eszköz megfelel az I. mellékletben említett, a biztonságosságra és a teljesítőképességre vonatkozó általános követelményeknek, kivéve a teljesítőképesség-vizsgálatban meghatározott szempontokat;
- a teljesítőképesség-vizsgálatokat úgy kell megtervezni és végrehajtani, hogy megvalósuljon és minden más érdekkel szemben elsőbbséget élvezzen az ilyen teljesítőképesség-vizsgálatokban részt vevő személyek jogainak, biztonságának, méltóságának és jóllétének a védelme, valamint, hogy a vizsgálatokból származó klinikai adatok tudományosan érvényesek, megbízhatók és megalapozottak legyenek.
A teljesítőképesség-vizsgálatokat – a maradványmintákat használó teljesítőképesség-vizsgálatokat is beleértve – az adatvédelemre vonatkozó hatályos jognak megfelelően kell végezni.
Az IVDR rendelet alkalmazásának időpontja 2022. május 26. Azon teljesítőképesség-vizsgálatok esetében, melyeket ezen időpont előtt benyújtottak, a 98/79/EK irányelv (IVDD) és az ezt honosító, az in vitro diagnosztikai orvostechnikai eszközökről szóló 8/2003. (III.13.) ESzCsM rendelet előírásai szerint folytathatók.
2022. május 26-át követően már csak az IVDR-nek megfelelően lehet teljesítőképesség vizsgálatot engedélyeztetni, lefolytatni.
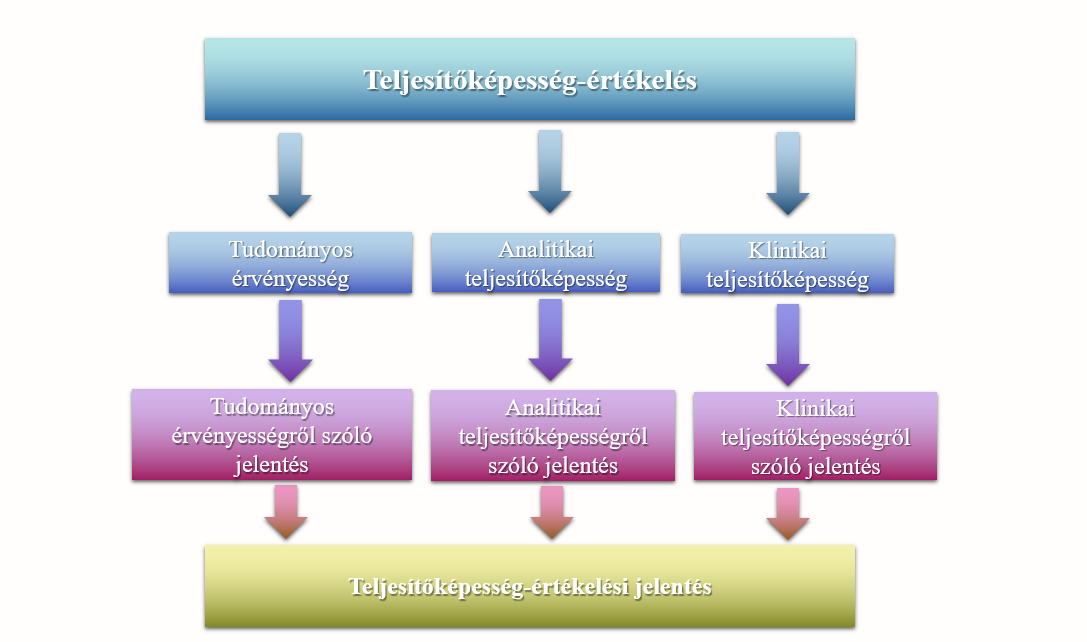
2. Teljesítőképesség-értékelés jól megalapozott (well-established) eszköz esetén
Amennyiben az eszköz régóta forgalomban van és a gyártónak minden szükséges információ rendelkezésre áll, akkor elkészíti az előbbiekben ismertetett teljesítőképesség-értékelési tervet, majd a tudományos érvényességről szóló jelentést, az analitikai teljesítőképességről szóló jelentést és a klinikai teljesítőképességről szóló jelentést, ezt követően pedig elkészíti – ezek összegzéseként – a teljesítőképesség-értékelési jelentést. A teljesítőképesség-értékelési dokumentációnak az adott eszköz műszaki dokumentációjának részét kell képeznie.
Az IVDR 56. cikkének (4) pontja értelmében a XIII. melléklet A. részének 2. pontja szerinti klinikai teljesítőképesség-vizsgálatok elvégzésétől csak abban az esetben lehet eltekinteni, ha a klinikai teljesítőképességre vonatkozó adatok egyéb forrásainak igénybevétele kellőképpen indokolt.
3. Hatósági bevonást igénylő teljesítőképesség-vizsgálatok
3.1. Kérelem/bejelentés benyújtásának feltételei és előzményei
- Amennyiben az adott teljesítőképesség-vizsgálat megbízója nincs letelepedve az Európai Unióban, gondoskodnia kell arról, hogy egy unión belül letelepedett természetes vagy jogi személy lássa el az unión belül a jogi képviseletét (IVDR 58. cikk (4) bekezdés).
- A Magyarország területén székhellyel rendelkező, illetve a Magyarország területén székhellyel rendelkező meghatalmazott képviselővel rendelkező gyártónak, a Magyarország területén székhellyel rendelkező meghatalmazott képviselőnek az IVDR 28. cikk szerinti regisztrációnak eleget kell tennie, SRN-számmal rendelkeznie kell. A klinikai vizsgálatra szánt eszköz gyártójának minden esetben rendelkeznie kell SRN számmal https://ogyei.gov.hu/eudamed_actor_modul
- A teljesítőképesség-vizsgálat megbízójának kérelmet/bejelentést kell benyújtania a klinikai vizsgálat végzésének helye szerinti tagállam orvostechnikai eszköz hatóságához (NNGYK).
- A kérelmet/bejelentést az IVDR 69. cikkben említett elektronikus rendszeren keresztül (EUDAMED klinikai vizsgálat modul) kell benyújtani. Ameddig az EUDAMED „klinikai vizsgálatok modul”-ja nem üzemel, addig az NNGYK a kérelmeket e-ügyintézés* keretében fogadja.
- Az adott teljesítőképesség-vizsgálatra/IVD eszközös vizsgálatra vonatkozóan Unió-szerte egységes, egyedi azonosító számot kell generálnia az EUDAMED rendszernek (CIV-ID), amelyet minden esetben meg kell adni az adott klinikai vizsgálattal kapcsolatos kommunikáció során. Ameddig az EUDAMED „klinikai vizsgálatok modul”-ja nem üzemel, addig az NNGYK az EUDAMED2 rendszerben nyilvántartásba veszi a klinikai vizsgálatot és az ott kapott azonosítót megküldi a megbízónak.
*Az elektronikus ügyintézés és a bizalmi szolgáltatások általános szabályairól szóló 2015. évi CCXXII. törvény (e-ügyintézési tv.) értelmében 2018. január 1-től kötelező a gazdálkodó szervezetek számára az egyes állami szervekkel való elektronikus kapcsolattartás, valamint az ehhez szükséges hivatalos elérhetőségen történő dokumentumküldés/fogadás. Hivatalos ügy indításához segédlet az alábbi linken található: https://ogyei.gov.hu/eugyintezes
3.1.1. Pozitív etikai vélemény szerzése
A teljesítőképesség-értékelési vizsgálat az Egészségügyi Tudományos Tanács Tudományos és Kutatásetikai Bizottság (ETT TUKEB) által - az (EU) 2017/746 rendelet 58. cikk (3) alapján - kiadott támogató etikai véleményének birtokában kezdeményezhető az NNGYK-nál.
Az etikai véleménnyel kapcsolatosan kérjük, keresse az Egészségügyi Tudományos Tanács Tudományos és Kutatásetikai Bizottságot (ETT TUKEB) az alábbi elérhetőségeken:
Levelezési cím: Belügyminisztérium, 1903 Budapest, Pf.: 314.
Telefon: (+36 1) 795-1197 (+36 1) 795-1198
E-mail: Ez az e-mail-cím a szpemrobotok elleni védelem alatt áll. Megtekintéséhez engedélyeznie kell a JavaScript használatát.
Honlap: https://ett.okfo.gov.hu/tukeb/
3.2. Bejelentésköteles teljesítőképesség vizsgálatok
3.2.1. Teljesítőképesség-vizsgálatok maradványminták felhasználásával
Az egészségügyről szóló 1997. évi CLIV. törvény 164/A. §. (10) pontja értelmében a bejelentésköteles teljesítőképesség-értékelési vizsgálatok a következők:
-
az (EU) 2017/746 rendelet 58. cikk (1) bekezdése alá nem tartozó, in vitro diagnosztikai orvostechnikai eszközökkel végzett teljesítőképesség-vizsgálatok, (nem történik a teljesítőképesség-vizsgálat céljából célzott mintavétel, vagyis a teljesítőképesség-vizsgálat maradékminták felhasználásával történik);
-
az (EU) 2017/746 rendelet 58. cikk (2) bekezdése szerinti, a kapcsolt diagnosztikumok kizárólag maradványminták felhasználásával végzett teljesítőképesség-vizsgálata.
Az NNGYK a bejelentés megtételét követő 60 napon belül hatósági bizonyítványt állít ki.
3.2.2. Teljesítőképesség-vizsgálatok CE-jelöléssel ellátott eszközök esetében
Az IVDR 70. cikk (1) bekezdése szerinti vizsgálat esetén a megbízónak legkésőbb 30 nappal a vizsgálat megkezdését megelőzően be kell jelentenie az NNGYK részére a teljesítőképesség-vizsgálatot az EUDAMED-en keresztül.
A vizsgálat a 1997. évi CLIV. törvény 164/A. §. (10) pontja értelmében a bejelentésköteles, azonban a kérelem benyújtása során a 2.6. pontban ismertetett eljárásrend követendő. Amennyiben a benyújtott bejelentés hiányos, csak a teljes kiegészítéstől számított 30. napon kezdhető meg a vizsgálat a nyilvántartásba vételi igazolás birtokában. Felhívjuk figyelmét, hogy az Eütv. lehetőséget ad az NNGYK-nak, hogy az igazolást a hiánytalan bejelentés időpontjától számított 30 naptári napon belül állítsa ki.
IVDR 70. cikk (2) bekezdése szerinti vizsgálat esetén a kérelem benyújtásának módja a mintavétel módjától függ.
3.3. Engedélyköteles teljesítőképesség-értékelési vizsgálatok
Az engedélyköteles vizsgálatok körét az emberen végzett orvostudományi kutatások, az emberi felhasználásra kerülő vizsgálati készítmények klinikai vizsgálata, valamint az emberen történő alkalmazásra szolgáló, klinikai vizsgálatra szánt orvostechnikai eszközök klinikai vizsgálata engedélyezési eljárásának szabályairól szóló 235/2009. (X.20.) Korm. rendelet határozza meg. A fenti rendelet 30.§ (1) pontja értelmében engedélyköteles vizsgálatok a következők:
-
az IVDR 58. cikk (1) bekezdése szerinti olyan teljesítőképesség-vizsgálatok,
-
amelyek során kizárólag a teljesítőképesség-vizsgálat céljából kerül sor sebészeti invazív mintavételre;
-
amelyek az IVDR 2. cikk 46. pontjában meghatározott, beavatkozással járó klinikai teljesítőképesség-vizsgálatok; vagy
-
amelyek során a vizsgálat további invazív eljárásokkal vagy a vizsgálati alanyokra nézve egyéb kockázatokkal jár,
-
az IVDR 58. cikk (2) bekezdése szerinti kapcsolt diagnosztikum nem maradványminták felhasználásával végzett vizsgálata,
-
IVDR 70. cikk (2) bekezdése szerinti teljesítőképesség-vizsgálatok (amennyiben célzott mintavétel történik).
Az IVDR 58. cikk (1) bekezdése szerinti teljesítőképesség-értékelési vizsgálat az ETT TUKEB bizottságának az (EU) 2017/746 rendelet 58. cikk (5) bekezdés b) pontja szerint kiadott támogató etikai véleményének birtokában kérelmezhető.
A vizsgálatot az NNGYK engedélyezi és nyilvántartásba veszi a 235/2009. (X.20.) Korm. rendelet 32.§-nak megfelelően.
A teljesítőképesség-vizsgálatot csak akkor lehet lefolytatni, ha az IVDR 58. cikk (5) pontjában felsorolt feltételek mindegyike teljesül.
3.4. A hatósági eljárás folyamata
Felhívjuk figyelmüket, hogy a kérelem befogadására és elbírálására az IVDR 66. cikkének rendelkezései az irányadók.
3.4.1. A kérelem validálása
-
Az a dátum, amikor a megbízót az NNGYK értesíti, hogy a kérelem az IVDR hatálya alá tartozik és a kérelem hiánytalan. A kérelem validálása után kezdi meg az NNGYK az IVDR 67. cikkében meghatározott értékelést, mely alatt a hatóság felszólíthatja a megbízót kiegészítő információk benyújtására.
Az NNGYK a validálás dátumától számított 45 napon belül – az értékelési eljárás végén – értesíti a megbízót az engedélyről. A tagállam szakértői konzultációk céljából további 20 nappal meghosszabbíthatja az értékelésre adott időtartamot.
A megbízó az alábbi körülmények között kezdheti el a teljesítőképesség vizsgálatot:
-
Az IVDR 58. cikk (1) bekezdésének a) pontja szerint végzett olyan teljesítőképesség-vizsgálat esetén, amely során a mintagyűjtés nem jár jelentős klinikai kockázattal a vizsgálati alanyokra nézve a vizsgálat a kérelem validálását követően kezdhető meg. (lsd. a példákat a 2. sz. mellékletben)
-
Az alábbi teljesítőképesség-vizsgálatokat csak az NNGYK által kiadott engedély véglegessé válását követően lehet megkezdeni:
-
az IVDR 58. cikk (1) bekezdése szerinti olyan teljesítőképesség-vizsgálatok,
-
amelyek során kizárólag a teljesítőképesség-vizsgálat céljából kerül sor sebészeti invazív mintavételre és ez jelentős kockázattal jár a vizsgálati alanyokra nézve, ld. 2. sz. melléklet;
-
amelyek az IVDR 2. cikk 46. pontjában meghatározott, beavatkozással járó klinikai teljesítőképesség-vizsgálatok; vagy
-
amelyek során a vizsgálat további invazív eljárásokkal vagy a vizsgálati alanyokra nézve egyéb kockázatokkal jár, (ld. a példákat a 2. sz. mellékletben);
-
az IVDR 58. cikk (2) bekezdése szerinti kapcsolt diagnosztikum nem maradványminták felhasználásával végzett vizsgálata.