
Guidance on the IVDR performance evaluation process
I. Definitions
in vitro diagnostic medical device means any medical device which is a reagent, reagent product, calibrator, control material, kit, instrument, apparatus, piece of equipment, software or system, whether used alone or in combination, intended by the manufacturer to be used in vitro for the examination of specimens, including blood and tissue donations, derived from the human body, solely or principally for the purpose of providing information on one or more of the following:
a) concerning a physiological or pathological process or state;
b) concerning congenital physical or mental impairments;
c) concerning the predisposition to a medical condition or a disease;
d) to determine the safety and compatibility with potential recipients;
e) to predict treatment response or reactions;
f) to define or monitoring therapeutic measures.
Specimen receptacles shall also be deemed to be in vitro diagnostic medical devices;
clinical evidence means clinical data and performance evaluation results, pertaining to a device of a sufficient amount and quality to allow a qualified assessment of whether the device is safe and achieves the intended clinical benefit(s), when used as intended by the manufacturer;
clinical benefit means the positive impact of a device related to its function, such as that of screening, monitoring, diagnosis or aid to diagnosis of patients, or a positive impact on patient management or public health;
scientific validity of an analyte means the association of an analyte with a clinical condition or a physiological state;
performance of a device means the ability of a device to achieve its intended purpose as claimed by the manufacturer. It consists of the analytical and, where applicable, the clinical performance supporting that intended purpose;
analytical performance means the ability of a device to correctly detect or measure a particular analyte;
clinical performance means the ability of a device to yield results that are correlated with a particular clinical condition or a physiological or pathological process or state in accordance with the target population and intended user;
performance study means a study undertaken to establish or confirm the analytical or clinical performance of a device;
performance study plan means a document that describes the rationale, objectives, design methodology, monitoring, statistical considerations, organisation and conduct of a performance study;
performance evaluation means an assessment and analysis of data to establish or verify the scientific validity, the analytical and, where applicable, the clinical performance of a device;
device for performance study means a device intended by the manufacturer to be used in a performance study. A device intended to be used for research purposes, without any medical objective, shall not be deemed to be a device for performance study;
interventional clinical performance study means a clinical performance study where the test results may influence patient management decisions and/or may be used to guide treatment;
subject means an individual who participates in a performance study and whose specimen(s) undergo in vitro examination by a device for performance study and/or by a device used for control purposes;
investigator means an individual responsible for the conduct of a performance study at a performance study site;
sponsor means any individual, company, institution or organisation which takes responsibility for the initiation, for the management and setting up of the financing of the performance study;
informed consent means a subject's free and voluntary expression of his or her willingness to participate in a particular performance study, after having been informed of all aspects of the performance study that are relevant to the subject's decision to participate or, in the case of minors and of incapacitated subjects, an authorisation or agreement from their legally designated representative to include them in the performance study;
ethics committee means an independent body established in a Member State in accordance with the law of that Member State and empowered to give opinions for the purposes of this Regulation, taking into account the views of laypersons, in particular patients or patients' organisations. In Hungary, this is the Scientific and Research Ethics Committee of the Scientific Council for Health (TUKEB). Its opinion must be attached to the application for an investigational medicinal product as a condition for the granting of an investigational medicinal product authorisation. For details on the application for an ethics opinion, the procedure for issuing an opinion and the fees, please visit the official website of the ETT TUKEB: https://ett.aeek.hu/tukeb/
adverse event means any untoward medical occurrence, inappropriate patient management decision, unintended disease or injury or any untoward clinical signs, including an abnormal laboratory finding, in subjects, users or other persons, in the context of a performance study, whether or not related to the device for performance study;
serious adverse event means any adverse event that led to any of the following:
a) a patient management decision resulting in death or an imminent life-threatening situation for the individual being tested, or in the death of the individual's offspring,
b) death,
c) serious deterioration in the health of the individual being tested or the recipient of tested donations or materials, that resulted in any of the following:
• life-threatening illness or injury,
• permanent impairment of a body structure or a body function,
• hospitalisation or prolongation of patient hospitalisation,
• medical or surgical intervention to prevent life-threatening illness or injury or permanent impairment to a body structure or a body function,
• chronic disease,
d) foetal distress, foetal death or a congenital physical or mental impairment or birth defect;
post-market surveillance means all activities carried out by manufacturers in cooperation with other economic operators to institute and keep up to date a systematic procedure to proactively collect and review experience gained from devices they place on the market, make available on the market or put into service for the purpose of identifying any need to immediately apply any necessary corrective or preventive actions;
incident means any malfunction or deterioration in the characteristics or performance of a device made available on the market, including use-error due to ergonomic features, as well as any inadequacy in the information supplied by the manufacturer and any harm as a consequence of a medical decision, action taken or not taken on the basis of information or result(s) provided by the device;
serious incident means any incident that directly or indirectly led, might have led or might lead to any of the following:
the death of a patient, user or other person,
the temporary or permanent serious deterioration of a patient's, user's or other person's state of health,
a serious public health threat;
serious public health threat means an event which could result in imminent risk of death, serious deterioration in a person's state of health, or serious illness, that may require prompt remedial action, and that may cause significant morbidity or mortality in humans, or that is unusual or unexpected for the given place and time.
II. Related legislation
Legislation related to the performance testing of IVDs:
- Act CLIV of 1997 on Health Care;
- EU Regulation 2017/746 on in vitro diagnostic medical devices (IVDR);
- Regulation (ECMR) No 8/2003 (13.3.2003) on in vitro diagnostic medical devices;
- Regulation (EC) No 33/2009 (X. 20.) on the clinical investigation of medical devices;
- Government Decree No 235/2009 (X. 20.) on the rules of the authorisation procedure for medical research involving human subjects, clinical trials of investigational medicinal products for human use and clinical trials of medical devices intended for clinical investigation in human subjects;
- Decree 23/2002 (9 May 2002) of the Ministry of Health on medical research on humans;
- Decree No 50/1996 (XII. 27.) NM on the fees for certain administrative procedures and services of an administrative nature in the field of public welfare.
III. General information
The performance evaluation of IVD devices is required for manufacturers by Article 56 of Regulation (EU) 2017/746 (hereinafter IVDR). The purpose of the performance evaluation is to provide sufficient clinical evidence of the compliance of the IVD device with the safety and performance requirements, and to assess interference, cross-reactivity and the acceptability of the risk-benefit balance associated with the use of the device.
The following must be confirmed during the performance evaluation study of a device:
• scientific validity;
• analytical performance;
• clinical performance.
The results of the assessment of the three elements provide the necessary clinical evidence.
The process for carrying out the performance evaluation
Performance evaluation is an ongoing activity following a documented performance evaluation plan. To implement it, the manufacturer shall prepare a performance evaluation plan in accordance with Annex XIII, point 1.1 of the IVDR. It shall then perform the verification of scientific validity (Annex IVDR XIII 1.2.1), the verification of analytical performance (Annex IVDR XIII 1.2.2) and the verification of clinical performance (Annex IVDR XIII 1.2.3). Each of the three steps shall be demonstrated in a single report. From these three reports, a Performance Evaluation Report (PER) (Annex IVDR XIII, Part A, point 1.3.2) must be produced. The process is illustrated in the figure below:
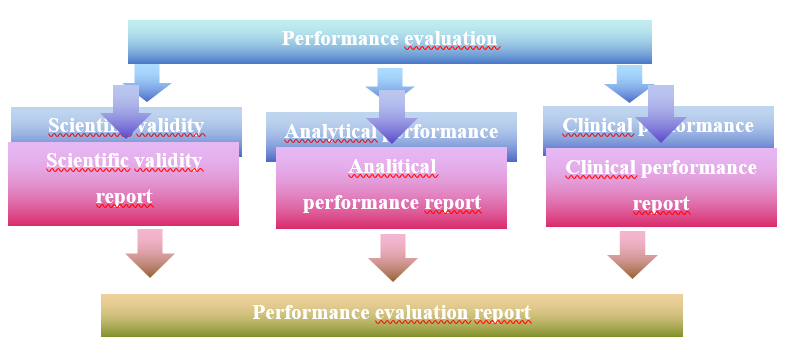
Figure 1. Process of the performance evaluation
The performance evaluation is part of the technical documentation.
The performance evaluation and its documentation must be updated throughout the life cycle of the device, based on the post-market surveillance system and the associated plan for its implementation (Articles 78-79 of the IVDR and Annex III, point 1).
A range of data can be collected through the post-market surveillance system, listed in Article 78(3) of the IVDR. Of particular importance may be the post-market surveillance of performance (as defined in Annex XIII, Part B) and the plan for this, which may include a post-market performance monitoring study (PMPF study).
The Performance Evaluation Report (PER) for Class C and D devices must be updated as necessary, but at least annually (Periodic Device Safety Report - IVDR Article 81). Similarly, for Class A and B devices, a post-market surveillance report must be prepared regularly for manufacturers (IVDR Article 80). In addition, for devices in Class C and D, a Safety and Performance Summary (SSCP) is required under Article 29 of the IVDR, which must be drafted in a way that is clear to the intended users and made publicly available through EUDAMED. Until the EUDAMED is operational, the manufacturer shall comply by publishing on its website or other publicly accessible web-based platform.
IV. Ways to carry out a performance evaluation
1. General requirements for performance evaluation studies
- performance evaluation studies shall be carried out under conditions similar to those under which the instrument is normally used;
- the manufacturer shall ensure that the device intended for performance evaluation complies with the general safety and performance requirements referred to in Annex I, except for the aspects specified in the performance test;
- performance evaluation studies are designed and conducted in such a way that the rights, safety, dignity and well-being of the persons participating in such performance evaluation studies are protected and take precedence over all other interests, and that the clinical data generated from such studies are scientifically valid, reliable and robust.
Performance evaluation studies, including performance evaluation studies using residual specimens, shall be conducted in accordance with the applicable data protection legislation.
The date of application of the IVDR Regulation is 26 May 2022. For performance studies submitted before this date, they may be conducted in accordance with the provisions of Directive 98/79/EC (IVDD) and its successor, the ECMR 8/2003 (13.3.2003) on in vitro diagnostic medical devices.
After 26 May 2022, performance testing can only be authorised and conducted in accordance with the IVDD.
2. Performance evaluation for a well-established device
If the device has been on the market for a long time and the manufacturer has all the necessary information, it will prepare the performance evaluation plan described above, followed by the scientific validity report, the analytical performance report and the clinical performance report, and then, summarising these, the performance evaluation report. The performance evaluation documentation shall be part of the technical documentation of the device.
According to Article 56(4) of the IVDR, the clinical performance tests under Annex XIII, Part A, point 2, may be waived only if there is sufficient justification for using other sources of clinical performance data.
3. Performance evaluation studies requiring regulatory involvement
3.1 Conditions and history of the application/notification
- If the sponsor of the performance evaluation is not established in the Union, it must ensure that a natural or legal person established in the Union acts as its legal representative in the Union (Article 58(4) of the IVDR).
- A manufacturer established in Hungary or having an authorised representative established in Hungary, an authorised representative established in Hungary must be registered in accordance with Article 28 of the IVDR and have an SRN. In any case, the manufacturer of the device intended for clinical investigation must have an SRN https://ogyei.gov.hu/eudamed_actor_modul
- The sponsor of the performance study must submit an application/notification to the Medical Device Authority (MDA) of the Member State where the clinical trial is to be conducted.
- The application/notification must be submitted via the electronic system referred to in Article 69 of the IVDR (EUDAMED clinical trial module). As long as the EUDAMED "clinical trials module" is not operational, the NNGYK will accept applications via e-Administration*.
- For a given performance study/IVD device trial, a unique EU-wide identification number (CIV-ID) must be generated by the EUDAMED system and must be provided in all communications related to the clinical trial. As long as the EUDAMED "clinical trials module" is not operational, the NNGYK will register the clinical trial in EUDAMED2 and send the ID received there to the sponsor.
*According to Act CCXXII of 2015 on the General Rules of Electronic Administration and Trust Services (e-Administration Act), from 1 January 2018, it is mandatory for business organisations to communicate electronically with certain public authorities and to send/receive documents via the official contact details required for this purpose. Help for starting an official case can be found at the following link: https://ogyei.gov.hu/eugyintezes
3.1.1. Obtaining a positive ethical opinion
The performance evaluation studies can be initiated with the support of the Scientific and Research Ethics Committee of the Scientific Council for Health (ETT TUKEB), in the possession of a favourable ethical opinion issued by the Scientific Committee of the Scientific Council of the Health Sciences (SCS), pursuant to Article 58(3) of Regulation (EU) 2017/746.
For the ethical opinion, please contact the Scientific and Research Ethics Committee of the Scientific Council for Health (ETT TUKEB) at the following contact details:
Ministry of the Interior, 1903 Budapest, PO Box 314.
Telephone: (+36 1) 795-1197 (+36 1) 795-1198
E-mail:
Website: https://ett.okfo.gov.hu/tukeb/
3.2. Notifiable performance evaluation study
3.2.1. Performance evaluation studies using leftover samples
Pursuant to Article 164/A(10) of Act CLIV of 1997 on Health Care, the following performance evaluation studies are notifiable:
• performance tests carried out with in vitro diagnostic medical devices not covered by Article 58(1) of Regulation (EU) 2017/746, (no targeted sampling for performance evaluation purposes, i.e. performance testing is carried out using residual samples)
• performance evaluation of companion diagnostic tools using only leftover samples, as referred to in Article 58(2) of Regulation (EU) 2017/746,
The NNGYK shall issue an official certificate within 60 days of the notification.
3.2.2. Performance evaluation studies for devices bearing the CE marking
In the case of a preformance evaluation study under Article 70(1) of the IVDR, the sponsor must notify the NNGYK of the performance study via EUDAMED at least 30 days before the start of the study.
The performance evaluation study is notifiable pursuant to Article 164/A(10) of Act CLIV of 1997, but the procedures described in point 2.6 must be followed when submitting the application. If the notification submitted is incomplete, the examination may only be started on the 30th day after the complete notification has been submitted and a certificate of registration has been obtained. Please note that the Health Act allows the NNGYK to issue the certificate within 30 calendar days from the date of the complete notification.
In the case of an investigation under Article 70(2) of the IVDR, the way in which the application is submitted depends on the method of sampling.
3.2. Performance evaluation studies requiring an approval
The scope of the studies requiring authorisation is defined by Government Decree 235/2009 (X.20.) on the rules of the authorisation procedure for medical studies in humans, clinical studies of investigational medicinal products for human use and clinical studies of medical devices intended for clinical studies in humans. Pursuant to Article 30(1) of the above Regulation, the following studies are subject to authorisation:
a) performance tests as referred to in Article 58(1) of the IVDR,
• which involve surgical invasive sampling for the sole purpose of performance testing;
• interventional clinical proficiency testing as defined in Article 2(46) of the IVDR; or
• which involve additional invasive procedures or other risks to subjects,
b) the testing of a linked diagnostic test using non-leftover specimens as defined in Article 58(2) of the IVDR,
c) performance testing under Article 70(2) IVDR (where targeted sampling is used)
The performance assessment test under Article 58(1) of the IVDR may be requested in the possession of a supporting ethical opinion issued by the ETF TUKEB Committee in accordance with Article 58(5)(b) of Regulation (EU) 2017/746.
The examination is authorised and registered by the NNGYK in accordance with § 32 of Government Decree No 235/2009 (X.20.).
The performance test may only be conducted if all the conditions listed in Article 58(5) of the IVDR are fulfilled.
3.4. The administrative procedure
Please note that the provisions of Article 66 of the IVDR apply to the reception and processing of your application.
• Within 10 days of receipt of a request for a performance evaluation study, the NNGYK will notify the sponsor (this period may be extended by 5 days by the NNGYK) whether the performance test is covered by the IVDR and whether the request received is complete.
• If the NNGYK determines that the proficiency testing is not covered by the IVDR or the application is incomplete, it will inform the sponsor and set a 10-day deadline (which may be extended by the NNGYK by 20 days upon request) for the sponsor to comment on or complete the application. Comments and additions must be made via EUDAMED/e-administration.
• If the sponsor has not commented and/or completed the application within the above time limit, the application will be deemed to be terminated (a termination order will be sent).
• If the sponsor considers that the application is covered by the IVDR and/or that the application is complete, but the NNGYK disagrees, the application will be considered as rejected (a termination order will be issued). The NNGYK will provide an appeal procedure in case of termination.
• If the sponsor has submitted comments or additions within the above time limit, the NNGYK will notify the sponsor within 5 days of receipt (this time limit may be extended by 5 days by the NNGYK) whether the performance test is covered by the IVDR and whether the application received is complete (this is the notification of validation of the application.)
• In all cases, deadlines are counted in calendar days!
3.4.1. Validation of the application
• The date on which the sponsor is notified by the NNGYK that the application is subject to the IVDR and that the application is complete.
• After the application has been validated, the NNGYK will start the assessment as defined in Article 67 of the IVDR, during which the Authority may request the sponsor to submit additional information. The NNGYK will notify the sponsor of the authorisation within 45 days of the date of validation, at the end of the assessment procedure. The Member State may extend the evaluation period by a further 20 days for expert consultations.
The sponsor may start the performance assessment under the following circumstances:
• In the case of a proficiency test conducted in accordance with Article 58(1)(a) of the IVDR where the collection of the specimen does not pose a significant clinical risk to the subjects, the test may be initiated after the validation of the application (see examples in Annex 2)
• The following performance studies may only be initiated after final approval by the NNGYF:
a) performance tests according to Article 58(1) of the IVDR,
b) which involve surgical invasive sampling for the sole purpose of performance testing and which pose a significant risk to subjects; see Annex 2.
c) which are interventional clinical performance studies as defined in Article 2(46) of the IVDR; or
d) where the study involves additional invasive procedures or other risks to subjects (see examples in Annex 2)
• testing of a companion diagnostic study using non-leftover samples, in accordance with Article 58(2) of the IVDR
V. List of documents to be submitted
Please submit the documents in one folder, zipped and numbered according to the Attachments/Annexed document!
Documents Articles 58 and 70 (2) IVDR
(approval) Article 70 (1) IVDR for devices with CE marking For leftover samples CLIV Act of 1997, § 164/A.(10). (Hungarian national law) Comments
Mellékletek/Annexes documentum + + + Find it on our website!
https://ogyei.gov.hu/dynamic/mellekletek_annexes.docx
Cover letter + + + a free-word letter addressed to the Medical Technology Department of the NNGYK requesting that the documents submitted be evaluated
Proof of payment of the administrative service fee + + +
Full list of devices used in the performance evaluation in Excel format + + +
Authorisation of the EU legal representative if the sponsor is established outside the EU + + + if the sponsor is not based in the EU
Performance evaluation study notification/application form + + + Find it on our website!; MDCG 2022-19
https://ogyei.gov.hu/dynamic/performance_study_application_form_hu.pdf
Performance Evaluation Plan (PEP) + + + IVDR Annex XIII Part A 1.1 (plan for scientific validity, analytical performance, clinical performance); MDCG 2022-2
Clinical performance study plan (CPSP) + + + According to Annex IVDR XIII Part A 2.3.2.
Investigator's Brochure (IB) Contents: + + -
the identification and description of the device, including information on its intended use, risk classification and applicable classification rules in accordance with Annex VIII, as well as the design and manufacture of the device, and a reference to previous and similar generations of the device
the manufacturer's instructions for the installation, maintenance, hygiene and use, including storage and handling requirements, as well as the information to be provided on the label, where available, and the instructions for use which must accompany the device when it is placed on the market. The fiche shall also include information on the relevant training required;
• analytical performance;
• existing clinical data;
• benefit-risk analysis;
for devices incorporating tissues, cells and substances of human, animal or microbial origin, detailed information on the tissues, cells and substances and their compliance with relevant general safety and performance requirements, as well as specific risk management for tissues, cells and substances
a list detailing how the general safety and performance requirements have been met, including the standards and uniform specifications that have been applied in full or in part, and a description of the solutions adopted to meet the relevant general safety and performance requirements where these standards and uniform specifications have not been applied or have been applied only in part or are missing
a detailed description of the clinical procedures and diagnostic tests used during the performance test and, in particular, information on deviations from standard clinical practice.
The device general safety and performance requirements list (GSPR list) + + + see IVDR Annex XIV 4.1
Positive ethical opinion + + + https://ett.okfo.gov.hu/tukeb/
Proof of subjects' insurance or entitlement to compensation in case of injury + + - see IVDR Annex XIV 4.3
Documents to be used to obtain informed consent, including the patient information leaflet and the informed consent document + + - ICF (see IVDR Annex XIV 4.4)
Description of the measures taken to comply with the applicable rules on the protection and confidentiality of personal data (GDPR) + + + see IVDR Annex XIV 4.5
List of available technical documentation and declaration to the authority that it will provide the documentation on request + - - see IVDR Annex XIV 4.6
Instructions for Use (english & hungarian language) + + +
Manufacturer’s Declaration of Conformity (DoC) - + -
Labels (english & hungarian language) + + +
Valid CE certificate or registration document, depending on the class of the device - + -
Documentation of the comparator device (DoC, CE certificate, instruction manual) + + + reference device documentation
Post-market performance monitoring plan - + +
(For CE marked device studies!)
IVDR Annex XIII Part B
Decisions by other Member States + + +
CVs of investigators + + -
Helsinki Declaration + + -
Declaration of acceptance by the testing site + + -
Declaration of competence of the testing sites + + -
Financial and other arrangements + + +
VI. Modification of the performance evaluation studies
A modification of a performance evaluation study is considered a substantial modification if the modification is likely to have a significant impact on the safety, health or rights of subjects or on the reliability or robustness of the data obtained from the study.
A modification is considered a significant or substantial modification for performance studies under Article 58(1) of the IVDR (Article 35 of Government Decree No. 235/2009 (20.X.2009)) if:
a) the modification relates to the purpose, circumstances or mechanism of action of the device,
b) the modification may change the interpretation of the scientific documents supporting the conduct of the study,
c) the modification affects the Investigator’s Brochure,
d) the results of the study to date require a change to the written information; or
e) the modification may affect the safety, health or rights of subjects.
In the case of studies carried out with IVDs that are subject to notification, a modification of the study plan is deemed to be a substantial modification (Article 19 (2) of Government Decree No 235/2009) if:
a) the modification may affect the safety of the subjects,
b) the modification may change the interpretation of the scientific documents supporting the conduct of the non-interventional trial,
c) the modification affects the investigator's brochure,
d) the trial results to date require a change to the written protocol.
Please note that any changes and/or modifictions to the data contained in the decision authorising the clinical performance study or in the official certificate (e.g. study site, investigator, sponsor, number of subjects enrolled, trial duration, etc.) must also be reported to the NNGYK.
1. How and when to submit a modification
If the sponsor intends to modify the IVD performance evaluation study in such a way that it can be considered a substantial modification (Article 71 of the IVDR) or if it needs to update the relevant data within one week of any change to the documentation referred to in Annex XIV, it must clearly indicate the change to the documentation. In addition, it must notify the Member State(s) where the performance evaluation study is being or will be conducted of the reason(s) and nature of the change within one week via the EUDAMED 'clinical trials module'. As long as the EUDAMED 'clinical trials module' is not operational, the NNGYK will receive requests or notifications of amendments via e-Administration.
2. Modification application and documents to be submitted
As part of the notification, the sponsor must attach an updated version of the relevant documentation referred to in Annex XIV of the IVDR, together with the application/notification form available on the NNGYK website. The relevant documentation must clearly indicate the changes. In addition to the proof of payment of the administrative service fee, the ETT TUKEB ethical opinion shall be attached to the application/notification of amendment. In the case of a negative, non-supportive opinion of the Ethics Committee, no modification of the approval/registration may be made.
3. The official procedure for approving a modification
The NNGYK will consider any significant modification to a performance evaluation study in accordance with the procedure laid down in Article 67 of the IVDR.
4. When can the modification can be implemented?
The sponsor may implement the modificatons to the performance evaluation study 38 days after a complete notification in EUDAMED (currently e-Administration), unless:
- the Member State in which the performance evaluation study is being or will be conducted has notified the sponsor that it has refused the modification on the basis of Article 67(4) of the IVDR or on the grounds of public health, the safety or health of subjects and users or public order,
- or the Ethics Committee of the Member State concerned has issued an unfavourable opinion on a major variation to a performance test which, according to the law of that Member State, applies to the whole territory of that Member State.
The Member State(s) concerned by the variation request may extend the period of 38 days referred to in Article 71(3) of the IVDR for a further seven (7) days for the purpose of expert consultations. Please note that, under national legislation, the procedure for transposition of amendments by the authority may take up to 60 days.
VII. Fees for administrative procedures
Pursuant to § 164/B of Act CLIV of 1997 on Health Care (Act on Health Care), an administrative service fee is payable for the authorisation or notification procedure for performance evaluation studies using in vitro diagnostic medical devices. For all types of performance evaluation studies with in vitro diagnostic medical devices, proof of payment of the administrative service fee must be provided at the time of submission of the application/notification, which is a condition for the initiation of the official procedure; failure to pay the fee will result in the application/notification being rejected.
Authorisation of performance evaluation of an in vitro diagnostic medical device - not including performance evaluations pursuant to Article 160/A (10) of Act CLIV of 1997 on Health Care - and approval of substantial modifications to the study plan 243 000 HUF/ application
Notification of a performance studies with an in vitro diagnostic medical device and notification of a major or substantial modification to the protocol 99 000 HUF/ notification
Financial details for bank transfer:
Name: Nemzeti Népegészségügyi és Gyógyszerészeti Központ
Tax number: 15598787-2-43
Bank: Magyar Államkincstár
Bank headquarters: 1139 Budapest, Hungary, Váci út 71.
Bankszámlaszám: 10032000-00290438-00000000
IBAN: HU551003-2000-0029-0438-0000-0000
Please indicate the identifier or the title of the study in the message box next to the word "orvostechnika". Please also note that the NNGYK can only invoice the payer of the administrative service fee, if the NNGYK have the originator's details and email address.
VIII. Interruption and termination of a performance evaluation study
• Where the sponsor temporarily interrupts or prematurely terminates a performance evaluation study, it must notify the Member States where the temporary interruption or premature termination of the performance evaluation study took place within 15 days, via an electronic system.
• Where the sponsor has temporarily interrupted or prematurely permanently stopped a performance evaluation study for safety reasons, it must notify all Member States where the performance evaluation study is being conducted within 24 hours.
• The end of the performance evaluation study shall be considered to be the last examination of the last subject, unless another date has been specified in the performance evaluation study plan as the end of the performance evaluation study.
• The sponsor shall also notify each Member State in which the performance evaluation study has been conducted of the completion of the performance evaluation study in that Member State. The notification shall be sent within 15 days of the completion of the performance evaluation study in the Member State concerned.
• In the case of studies carried out in more than one Member State, the sponsor shall notify all Member States in which the performance evaluation study was carried out of the completion of the performance evaluation study in all Member States. The notification shall be made within 15 days of the completion of the performance evaluation study.
• The sponsor shall, within one year after the completion of the performance evaluation study or within three months after the end of the premature permanent termination or temporary interruption, irrespective of the outcome of the performance evaluation study, submit the performance evaluation study report referred to in Annex XIII, Part A, point 2.3.3 of the IVDR to the Member States in which the performance evaluation study was carried out.
• The performance evaluation study report shall be accompanied by a summary written in a way that is easily understandable for the target users. The sponsor shall submit both the report and the summary through the EUDAMED electronic system.
• If for scientific reasons it is not possible to submit the performance study report within one year of the completion of the study, it shall be submitted as soon as possible when available. In this case, the performance evaluation study plan referred to in Annex XIII, Part A, point 2.3.2 shall specify when the results of the performance evaluation study will be available and shall provide a justification.
• If the IVD device is not registered in accordance with Article 26 of the IVDR within one year of the date of recording of the summary and the performance study report in the electronic system, the summary and the report shall be made publicly available after that one-year period.
• As long as the EUDAMED "clinical trials module" is not operational, the NNGYK will receive these documents through eadministration. Please indicate the CIV-ID and the NNGYK file number in the cover letter and on the documents.
• The NNGYK will send the final report submitted to it in connection with the performance evaluation study of IVD devices to the ETT TUKEB.
• Please send the Performance Evaluation Report (PER) via e-Administration to the NNGYK with the content and in due time according to Article 73 of the IVDR. Please indicate in the subject line: „Notification of the completion of the performance evaluation, Case number issued by the NNGYK' (use the case number provided in the approval letter).
IX Recording and reporting of adverse events during the performance evaluation studies
The sponsor shall promptly report the following vigilance events in relation to performance evaluation studies under Article 76(2) of the IVDR to the Member States where the clinical trial is conducted:
- any serious adverse event that is causally related to the device, reference device or trial procedure, or for which such a causal relationship can reasonably be assumed;
- any device failure that could have led to a serious adverse event in the absence of appropriate action or intervention or under less favourable circumstances;
- any new finding relating to the events referred to in the previous two points.
These reports may be submitted by the sponsor via the electronic system EUDAMED 'clinical trial module' referred to in Article 69 of the MDR. For Hungary, these reports are expected to be sent by the NNGYK to
The sponsor must also report adverse events occurring during clinical trials in the Member States concerned where the trial is conducted and any events occurring in third countries (outside EU Member States) where a trial is conducted under the same protocol as the protocol for trials covered by the IVDR.
ATTENTION! In the case of the post-market monitoring of performance referred to in Article 70(1) of the IVDR, the occurrence reporting shall be carried out in accordance with Articles 82 to 85 and 86 of the IVDR. And it must be sent to the authority via the EUDAMED "vigilance module" electronic system referred to in Article 87 of the MDR. The NNGYK expects vigilance reports to be sent to
However, if a causal link between the serious adverse event and the preceding PMCF study procedure is established during a post-marketing monitoring of performance (PMCF/ and non-interventional) study, then Article 76 of the IVDR applies to the vigilance report and the report should be entered in the EUDAMED "clinical trial module" and the NNGYK will await the notification at
Information and forms for the reporting of adverse events involving CE marked medical devices (including IVD devices) are available on the NNGYK website at the links below:
https://ogyei.gov.hu/varatlan_esemeny_jelentes https://ogyei.gov.hu/varatlan_esemeny_jelentes_formanyomtatvanyok
Projektek


![]()
